aHUS
| Site: | EHC | Egyptian Health Council |
| Course: | Pediatrics Guidelines |
| Book: | aHUS |
| Printed by: | Guest user |
| Date: | Thursday, 6 August 2026, 7:51 AM |
Description
"last update: 1 July 2025" Download Guideline
- Executive summary
1- In patients with suspected atypical hemolytic uremic syndrome (aHUS), the diagnosis should be based on the presence of thrombotic microangiopathy (TMA), defined by the concurrent presence of all three of the following:
· Microangiopathic hemolytic anemia (MAHA)
· Thrombocytopenia
· Acute kidney injury (AKI) (Good practice statement)
2- We suggest defining MAHA as hemoglobin <10 g/dL with evidence of intravascular hemolysis such as elevated LDH, low/undetectable haptoglobin, or increased reticulocyte count. (Conditional recommendation)
3- We suggest that a schistocyte count ≥2% on a peripheral blood smear supports the diagnosis of MAHA. A count ≥1% should raise clinical suspicion of TMA and prompt further evaluation. (Conditional recommendation)
4- We suggest defining thrombocytopenia in suspected aHUS or other thrombotic microangiopathies as a platelet count of less than 150,000/mm³ or a decrease of 25% or more from baseline, even if the absolute value remains within the normal range. (Conditional recommendation)
5- We suggest defining acute kidney injury in suspected aHUS as a serum creatinine increase of ≥ 1.5 × the upper limit of normal for age and sex. (Conditional recommendation)
6- In patients with suspected aHUS, clinicians should consider urine output < 0.5 mL/kg/h for ≥ 6 hours as supportive of acute kidney injury. (Good practice statement)
7- Measurement of blood urea nitrogen (BUN) may
be useful in patients with suspected aHUS as part of routine kidney function
assessment. While BUN elevation is nonspecific, it can aid in the evaluation of
acute kidney injury (AKI) when considered with other TMA-related laboratory
markers such as serum creatinine and hemolysis parameters.
(Good practice statement)
8- In patients with suspected aHUS, a direct antiglobulin
(Coombs) test may help exclude autoimmune hemolytic anemia as a cause of
microangiopathic hemolysis. A negative result supports the diagnosis of
thrombotic microangiopathy, including aHUS.
(Good practice statement)
9- ANA testing may be performed in patients with
suspected aHUS to help exclude autoimmune conditions such as systemic lupus
erythematosus that can mimic aHUS.
(Good practice statement)
10- Fibrin degradation products (FDPs) or D-dimer levels may be measured to help exclude disseminated intravascular coagulation (DIC) in the differential diagnosis of aHUS. Normal or mildly elevated levels make DIC less likely and support the diagnosis of aHUS.(Good practice statement)
11- We recommend testing for Shiga toxin (Stx) using stool culture and/or ELISA/PCR in all patients with suspected aHUS, especially those with diarrheal prodrome. A positive result confirms STEC-HUS and excludes aHUS (Strong recommendation)
12- We recommend measuring ADAMTS13 activity and autoantibodies in all patients with suspected aHUS to exclude thrombotic thrombocytopenic purpura (TTP). A confirmed ADAMTS13 activity level <10% is diagnostic of TTP and excludes aHUS. Samples should be obtained prior to plasma exchange or transfusion to preserve diagnostic accuracy. (Strong recommendation)
13- We suggest measuring serum complement levels (C3 and C4) in all patients with suspected aHUS as part of the diagnostic workup. Low C3 or C4 levels support complement pathway involvement but are not diagnostic alone. (Conditional recommendation)
14- We suggest measuring anti–Factor H (CFH) autoantibodies in patients with suspected aHUS, particularly in cases without a clear secondary trigger. Detection of these autoantibodies supports the diagnosis of acquired complement-mediated aHUS and may guide targeted immunosuppressive management. (Conditional recommendation)
15- Anti–Factor H autoantibodies are more frequently observed in pediatric aHUS cases, and testing may have a higher diagnostic yield in this population. (Good practice statement)
16- We suggest performing a comprehensive genetic analysis in patients with confirmed or highly suspected atypical hemolytic uremic syndrome (aHUS), particularly in cases without an identified secondary trigger. This testing should evaluate genes encoding complement regulatory proteins such as CFH, CFI, CFB, MCP (CD46), C3, and others. Identification of pathogenic variants supports the diagnosis of complement-mediated aHUS and may inform prognosis and therapeutic decision-making. (Conditional recommendation)
17- Genetic testing is not mandatory to confirm the initial diagnosis of aHUS, but it can provide valuable information for determining patient management, prognosis, and guiding the duration and intensity of treatment, particularly with C5 inhibitors. (Good practice statement)
18- Blood samples for ADAMTS13 activity, anti–factor H antibodies, and reticulocyte count should be obtained before any plasma therapy or transfusion in suspected aHUS or TMA cases. This ensures accurate diagnostic interpretation and informs appropriate treatment. (Good practice statement)
19- We recommend to start Plasma Exchange (PEX) within 24 hours of clinical suspicion of TMA (Strong recommendation)
20- PEX is done using 1.5 plasma volume exchanges of pure FFP in a dose of 60–75 mg/kg/day for 5 consecutive days. (Good practice statement)
21- The tapering schedule is every alternate day for 2 weeks, then twice weekly for 2 more weeks and eventually once weekly for an additional 4 weeks (Good practice statement)
22- Once the diagnosis of aHUS is confirmed, we recommend to use anti-C5 therapy as first line treatment (Strong recommendation)
23- : Use the following table for dosing (Table 1):
Table 1: Ravulizumab weight-based dosing regimen for pediatric patients with PNH or aHUS below 40 Kg
|
Body weight range (Kg) |
Loading dose (mg) |
Maintenance dose (mg)* |
Dosing interval |
|
≥ 10 to < 20 |
600 |
600 |
Every 4 weeks |
|
≥ 20 to < 30 |
900 |
2,100 |
Every 8 weeks |
|
≥ 30 to < 40 |
1,200 |
2,700 |
Every 8 weeks |
* First maintenance dose is administered 2 weeks after loading dose (Good practice statement)
24- Meningococcal vaccine should be given to reduce the infectious risk when anti-C5 therapy is initiated. (Good practice statement)
25- Patients treated with anti-C5 therapy <2 weeks after vaccination should be given daily prophylactic antibiotics for 2 weeks after vaccination. (Good practice statement)
In Patients positive for anti-CFH antibodies:
26- -In case of patients with positive anti-CFH antibodies, we recommend to start initial treatment by combining PEX and/or anti-C5 therapy with corticosteroids, with or without additional immunosuppressive agents (Strong recommendation)
27- Upon the remission of TMA, maintenance therapy is suggested to use corticosteroids ± immunosuppressants (Good practice statement)
28- If follow-up tests show anti-CFH antibodies are absent, discontinue immunosuppressive therapy (Good practice statement)
In patients negative for anti-CFH antibodies:
29- We recommend to continue treatment with anti-C5 therapy if genetic mutations identified (CFH, CFI, CFB, or C3 genes) is detected; discontinuation is not recommended as relapses are common post-cessation agents (Strong recommendation)
30- We suggest to use eculizumab in patients with atypical hemolytic uremic syndrome (aHUS), particularly in the following cases: after kidney transplantation to prevent or treat post-transplant thrombotic microangiopathy (TMA); in patients with active aHUS to improve renal outcomes and prevent dialysis; during perioperative periods to maintain complement inhibition and reduce surgical complications; and in combined organ transplantation to minimize the risk of graft thrombosis and optimize transplant success (Conditional recommendation)
- Recommendations
1. In patients with suspected atypical hemolytic uremic syndrome (aHUS), the diagnosis should be based on the presence of thrombotic microangiopathy (TMA), defined by the concurrent presence of all three of the following:
· Microangiopathic hemolytic anemia (MAHA)
· Thrombocytopenia
· Acute kidney injury (AKI) (Good practice statement) [12,14–16]
2. We suggest defining MAHA as hemoglobin <10 g/dL with evidence of intravascular hemolysis such as elevated LDH, low/undetectable haptoglobin, or increased reticulocyte count. (Conditional recommendation with moderate grade of evidence) [15,17,18]
3. We suggest that a schistocyte count ≥2% on a peripheral blood smear supports the diagnosis of MAHA. A count ≥1% should raise clinical suspicion of TMA and prompt further evaluation. (Conditional recommendation with moderate grade of evidence) [19–21]
4. We suggest defining thrombocytopenia in suspected aHUS or other thrombotic microangiopathies as a platelet count of less than 150,000/mm³ or a decrease of 25% or more from baseline, even if the absolute value remains within the normal range. (Conditional recommendation with moderate grade of evidence) [17,18,22,23]
5. We suggest defining acute kidney injury in suspected aHUS as a serum creatinine increase of ≥ 1.5 × the upper limit of normal for age and sex. (Conditional recommendation with moderate grade of evidence) [24,25]
6. In patients with suspected aHUS, clinicians should consider urine output < 0.5 mL/kg/h for ≥ 6 hours as supportive of acute kidney injury. (Good practice statement)
7. Measurement of blood urea nitrogen (BUN) may be useful in patients with suspected aHUS as part of routine kidney function assessment. While BUN elevation is nonspecific, it can aid in the evaluation of acute kidney injury (AKI) when considered with other TMA-related laboratory markers such as serum creatinine and hemolysis parameters. (Good practice statement)
8. In patients with suspected aHUS, a direct antiglobulin (Coombs) test may help exclude autoimmune hemolytic anemia as a cause of microangiopathic hemolysis. A negative result supports the diagnosis of thrombotic microangiopathy, including aHUS. (Good practice statement)
9. ANA testing may be performed in patients with suspected aHUS to help exclude autoimmune conditions such as systemic lupus erythematosus that can mimic aHUS. (Good practice statement)
10. Fibrin degradation products (FDPs) or D-dimer levels may be measured to help exclude disseminated intravascular coagulation (DIC) in the differential diagnosis of aHUS. Normal or mildly elevated levels make DIC less likely and support the diagnosis of aHUS. (Good practice statement)
11. We recommend testing for Shiga toxin (Stx) using stool culture and/or ELISA/PCR in all patients with suspected aHUS, especially those with diarrheal prodrome. A positive result confirms STEC-HUS and excludes aHUS (Strong recommendation with moderate grade of evidence) [14,15,18,22,24–26]
12. We recommend measuring ADAMTS13 activity and autoantibodies in all patients with suspected aHUS to exclude thrombotic thrombocytopenic purpura (TTP). A confirmed ADAMTS13 activity level <10% is diagnostic of TTP and excludes aHUS.
Samples should be obtained prior to plasma exchange or transfusion to preserve diagnostic accuracy. (Strong recommendation with moderate grade of evidence) [12,14,15,18,22,24–26]
13. We suggest measuring serum complement levels (C3 and C4) in all patients with suspected aHUS as part of the diagnostic workup. Low C3 or C4 levels support complement pathway involvement but are not diagnostic alone. (Conditional recommendation with moderate grade of evidence) [12–15]
14. We suggest measuring anti–Factor H (CFH) autoantibodies in patients with suspected aHUS, particularly in cases without a clear secondary trigger. Detection of these autoantibodies supports the diagnosis of acquired complement-mediated aHUS and may guide targeted immunosuppressive management. (Conditional recommendation with moderate grade of evidence) [11,13,18,24,27–32]
15. Anti–Factor H autoantibodies are more frequently observed in pediatric aHUS cases, and testing may have a higher diagnostic yield in this population. (Good practice statement)
16. We suggest performing a comprehensive genetic analysis in patients with confirmed or highly suspected atypical hemolytic uremic syndrome (aHUS), particularly in cases without an identified secondary trigger. This testing should evaluate genes encoding complement regulatory proteins such as CFH, CFI, CFB, MCP (CD46), C3, and others. Identification of pathogenic variants supports the diagnosis of complement-mediated aHUS and may inform prognosis and therapeutic decision-making. (Conditional recommendation with moderate grade of evidence) [13,27,33–35]
17. Genetic testing is not mandatory to confirm the initial diagnosis of aHUS, but it can provide valuable information for determining patient management, prognosis, and guiding the duration and intensity of treatment, particularly with C5 inhibitors. (Good practice statement)
18. Blood samples for ADAMTS13 activity, anti–factor H antibodies, and reticulocyte count should be obtained before any plasma therapy or transfusion in suspected aHUS or TMA cases. This ensures accurate diagnostic interpretation and informs appropriate treatment. (Good practice statement)
19. We recommend to start Plasma Exchange (PEX) within 24 hours of clinical suspicion of TMA (Strong recommendation with moderate grade of evidence) [1,35,36]
20. PEX is done using 1.5 plasma volume exchanges of pure FFP in a dose of 60–75 mg/kg/day for 5 consecutive days. (Good practice statement)
21. The tapering schedule is every alternate day for 2 weeks, then twice weekly for 2 more weeks, and eventually once weekly for an additional 4 weeks. (Good practice statement)
22. Once the diagnosis of aHUS is confirmed, we recommend to use anti-C5 therapy as first-line treatment (Strong recommendation with moderate grade of evidence) [1,12,35,37]
23. Use the following table for dosing (Table 4):
Table 4: Ravulizumab weight-based dosing regimen for pediatric patients with PNH or aHUS below 40 Kg
|
Body weight range (Kg) |
Loading dose (mg) |
Maintenance dose (mg)* |
Dosing interval |
|
≥ 10 to < 20 |
600 |
600 |
Every 4 weeks |
|
≥ 20 to < 30 |
900 |
2,100 |
Every 8 weeks |
|
≥ 30 to < 40 |
1,200 |
2,700 |
Every 8 weeks |
* First maintenance dose is administered 2 weeks after loading dose (Good practice statement)
24. Meningococcal vaccine should be given to reduce the infectious risk when anti-C5 therapy is initiated. (Good practice statement)
25. Patients treated with anti-C5 therapy <2 weeks after vaccination should be given daily prophylactic antibiotics for 2 weeks after vaccination. (Good practice statement)
In Patients positive for anti-CFH antibodies:
26. In case of patients with positive anti-CFH antibodies, we recommend to start initial treatment by combining PEX and/or anti-C5 therapy with corticosteroids , with or without additional immunosuppressive agents (Strong recommendation with moderate grade of evidence) [1,27,38–41]
27. Upon the remission of TMA, maintenance therapy is suggested to use corticosteroids ± immunosuppressants. (Good practice statement)
28. If follow-up tests show anti-CFH antibodies are absent, discontinue immunosuppressive therapy. (Good practice statement)
In patients negative for anti-CFH antibodies:
29. We recommend to continue treatment with anti-C5 therapy if genetic mutations identified (CFH, CFI, CFB, or C3 genes) is detected discontinuation is not recommended as relapses are common post-cessation agents (Strong recommendation with moderate grade of evidence) [6,42–44]
30. We suggest to use eculizumab in patients with atypical hemolytic uremic syndrome (aHUS), particularly in the following cases: after kidney transplantation to prevent or treat post-transplant thrombotic microangiopathy (TMA); in patients with active aHUS to improve renal outcomes and prevent dialysis; during perioperative periods to maintain complement inhibition and reduce surgical complications; and in combined organ transplantation to minimize the risk of graft thrombosis and optimize transplant success (Conditional recommendation with moderate grade of evidence) [45–49]
- Acknowledgement
Head of pediatric Supreme Committee for Guidelines of Egyptian Health Council:
General Doctor. Mourad Alfy Ramzy Tadros, * MD, FRCPCH(UK), MRCPI(Dublin)
* Consultant Paediatrician of Egyptian Military Medical Services.
* Professors of Paediatrics Military Medical Academy
* Head of Training Committee of Paediatrics of Egyptian Military Medical Board
* Consultant Paediatrician of the Medical Advisory Council of Egypt Healthcare
Authority (EHA).
Chair of Committee: Prof. Dr. Fatina Fadel
(Professor of pediatrics and pediatric nephrology – Faculty of Medicine – Cairo University)
Moderator of committee: Dr. Sara Abu El Enein
(Consultant of pediatrics and pediatric nephrology, Health Insurance Organization)
Members of Committee:
- Prof. Dr. Hafez Bazaraa:
Professor of Pediatrics, Head of PICU, Cairo University.
Pediatrics, Armed Forces College of Medicine. - Prof. Dr. Maher Abd El Hafez:
Professor of Pediatrics, Faculty of Medicine, Tanta University - Prof. Dr. Samar Sabry:
Professor of Pediatrics, Faculty of Medicine, Cairo University. - Prof. Dr. Rasha Esam:
Professor of Pediatrics, Faculty of Medicine, Cairo University. - Prof. Dr. Doaa Yousef:
Professor of Pediatrics, Faculty of Medicine, Elzagaziq University,
Head of Pediatric Department, Suez University - Dr. Ragia Marei:
Assistant Professor of Pediatrics, Faculty of Medicine, Ain Shams University - Dr. Ahmed Hussein:
Assistant Professor of Pediatrics, Faculty of Medicine, Ain Shams University
- List abbreviation
|
aHUS AKI |
Atypical hemolytic uremic syndrome Acute kidney injury |
|
anti-CFH |
Anti–complement factor H |
|
ANA |
Antinuclear antibody |
|
BUN |
Blood urea nitrogen |
|
C3 |
Complement 3 |
|
C5 CBC |
Complement 5 Complete blood count |
|
CFB |
Complement factor B |
|
CFH |
Complement factor H |
|
CFI CRP |
Complement factor I C-reactive protein |
|
DIC ESR |
Disseminated intravascular coagulation Erythrocytes sedimentation rate |
|
FFP |
Fresh frozen plasma |
|
FDPs |
Fibrin degradation products |
|
LDH MAHA |
Lactate dehydrogenase Microangiopathic Hemolytic Anemia |
|
MCP |
Membrane cofactor protein |
|
PEX |
Plasma exchange |
|
STEC-HUS Stx |
Shiga toxin–producing Escherichia coli–associated HUS Shiga toxin |
|
Stx ELISA |
Shiga toxin using enzyme-linked immunosorbent assay |
|
THBD |
Thrombomodulin |
|
TMA |
Thrombotic microangiopathy |
|
TTP |
Thrombocytopenic purpura |
- Glossary
A
ADAMTS13
A metalloprotease enzyme that cleaves von Willebrand factor; deficiency is
diagnostic for TTP
(thrombotic thrombocytopenic purpura), not aHUS.
Alternative Complement Pathway
A part of the innate immune system that activates complement independently of
antibodies. Dysregulation is central to aHUS pathophysiology.
Anti-CFH Antibodies
Autoantibodies against complement factor H, leading to uncontrolled complement
activation and TMA; often seen in pediatric aHUS.
Autoimmune aHUS
Subtype of aHUS involving anti-CFH antibodies, requiring immunosuppressive
treatment in addition to complement inhibition.
B
Blood Urea Nitrogen (BUN)
A test used alongside serum creatinine to assess kidney function.
C
CFH,
CFI, CFB, MCP, C3, THBD
Genes coding for regulatory proteins in the complement system. Mutations in
these are linked to genetic forms of aHUS.
Clinical Triad (aHUS)
Includes microangiopathic
hemolytic anemia, thrombocytopenia, and acute kidney injury.
Complement Inhibitors
Therapies (e.g., eculizumab, ravulizumab) that block C5 and prevent terminal
complement activation in aHUS.
Complement Levels (C3, C4)
Used to evaluate complement activity; low levels may indicate activation but
are not specific to aHUS.
C5b-9
(Soluble)
Terminal complement complex measured to assess complement activation; may
support aHUS diagnosis and monitoring.
D
Differential Diagnosis
The process of distinguishing aHUS from other TMAs such as TTP, STEC-HUS,
SLE, and DIC.
Direct Coombs Test
Used to differentiate immune hemolytic anemia from microangiopathic processes
like aHUS.
E
Eculizumab
A C5 inhibitor used in the treatment of aHUS; short-acting and administered
biweekly.
Endothelial Cell Dysfunction
A result of complement dysregulation in aHUS that leads to microvascular
thrombosis.
F
Familial aHUS
aHUS occurring in two or more related individuals, usually with inherited
complement gene mutations.
Fibrin Degradation Products (FDPs)
Used to rule out DIC in the differential diagnosis of TMA.
Full Fresh Frozen Plasma (FFP)
Used in plasma
exchange (PEX)
as replacement fluid
during the treatment of TMA.
G
Genetic Testing
Testing for mutations in complement genes to support diagnosis and guide
long-term therapy decisions in aHUS.
Glomerular Basement Membrane
A part of the kidney filtration barrier often damaged in aHUS due to
microthrombi and endothelial injury.
H
Hematuria
Presence of blood in urine, often a sign of kidney involvement in aHUS.
Hemolysis
Destruction of red blood cells seen in microangiopathic processes like aHUS.
Haptoglobin
A protein that binds free hemoglobin; low levels suggest hemolysis.
I
Immunosuppressive Therapy
Used in autoimmune aHUS (e.g., with anti-CFH antibodies) along with
corticosteroids and complement inhibitors.
K
Kidney Ultrasound
Imaging used to evaluate kidney structure in patients with suspected or ongoing
renal dysfunction.
L
Lactate Dehydrogenase (LDH)
An enzyme elevated in hemolysis; used to monitor disease activity in aHUS.
M
Maintenance Therapy
Continued treatment (e.g., anti-C5, immunosuppressants) after achieving
remission to prevent relapse in aHUS.
Microangiopathic Hemolytic Anemia (MAHA)
A form of hemolysis due to red cell destruction in the microvasculature; a
hallmark of aHUS.
Membrane Cofactor Protein (MCP/CD46)
A surface complement regulator; mutations are implicated in some genetic forms
of aHUS.
O
Oliguria
Decreased urine output; a sign of acute kidney injury, often seen in aHUS.
P
Plasma Exchange (PEX)
A therapeutic procedure replacing plasma to remove antibodies or abnormal complement
components; used in some aHUS cases.
Peripheral Blood Smear
Microscopic exam of blood cells; used to detect schistocytes in aHUS diagnosis.
PNH (Paroxysmal Nocturnal Hemoglobinuria)
Another complement-mediated condition treated with anti-C5 inhibitors, often
included in treatment studies alongside aHUS.
R
Ravulizumab
A long-acting C5 inhibitor administered every 8 weeks; used as an alternative
to eculizumab.
Reticulocyte Count
Used to assess bone marrow response to anemia; increased in hemolytic anemias.
S
Schistocytes
Fragmented red cells on blood smear, indicating microangiopathic hemolysis.
Shiga Toxin–Producing E. coli (STEC-HUS)
A common cause of typical HUS, which must be ruled out in suspected aHUS cases.
Systemic Lupus Erythematosus (SLE)
An autoimmune disease that can mimic TMA; must be excluded during aHUS workup.
T
Thrombotic Microangiopathy (TMA)
A group of disorders, including aHUS, marked by thrombosis in small vessels,
hemolysis, and thrombocytopenia.
Thrombocytopenia
Low platelet count, one component of the aHUS triad.
Thrombomodulin (THBD)
A complement-regulating protein; mutations can be associated with aHUS.
TTP (Thrombotic Thrombocytopenic Purpura)
A TMA condition that must be ruled out by ADAMTS13 testing in suspected aHUS
cases.
U
Uremia
A clinical syndrome of kidney failure with retention of waste products, common
in advanced aHUS.
- Introduction
Unlike typical HUS, which is often associated with Shiga toxin-producing Escherichia coli infections, aHUS is primarily linked to dysregulation of the alternative complement pathway [1]. This uncontrolled activation leads to systemic thrombotic microangiopathy (TMA), resulting in widespread microvascular thrombosis and subsequent organ damage, particularly in the kidneys [2]. Kidneys are the main target organs of HUS, with fibrin and platelet thrombi in glomerular capillaries, endothelial cell swelling, detachment from the glomerular basement membrane, and consequent severe renal impairment [3].
The etiology of aHUS is multifactorial, involving both genetic and acquired factors. Genetic dysregulation of the alternative complement pathway, resulting in endothelial cell dysfunction and microvascular thrombi formation, can be identified in 40–60% of aHUS patients [4]. This dysregulation follows mutations in genes coding for complement regulatory proteins, such as factor H (CFH), factor I (CFI), membrane cofactor protein (MCP), complement 3 (C3), factor B (CFB), or thrombomodulin (THBD), or by the presence of anti–complement factor H (anti-CFH) antibodies with consequent hyperactivation of the complement system [5].
Genetically determined aHUS can be familial or sporadic. The familial form represents about 20% of cases and it is defined by at least two members of the same family being diagnosed with aHUS in a maximum period of 6 months [6]. Genetic mutations predisposing to aHUS can be inherited in either an autosomal dominant or autosomal recessive manner or, rarely, as polygenic inheritance [6].
The development of aHUS is multifactorial. In fact, a second hit is necessary for the disease to manifest itself: different triggers are associated such as drugs (cisplatin, gemcitabine, mitomycin, clopidogrel, quinine, interferon-alfa/beta, anti-vascular endothelial growth factor, alemtuzumab, cyclosporin tacrolimus, ciprofloxacin, oral contraceptives, and vaccines) [6], infections (such as influenza, SARS-CoV-2), malignant hypertension, and pregnancy [7]. All these factors can lead to an onset of aHUS in genetically predisposed individuals. Generally, mutations affect a single gene; however, the combination of two or more mutations has been described in about 12% of patients [6].
Atypical hemolytic uremic syndrome (aHUS) is classified as an ultra-rare disease. Estimates suggest an annual incidence ranging from 0.23 to 1.9 cases per million people globally [8]. The prevalence is challenging to ascertain due to underdiagnosis and misclassification; however, some studies estimate it to be approximately 2 to 9 cases per million population. These figures underscore the rarity of the condition and the necessity for heightened clinical awareness.
The clinical burden of aHUS is substantial. Patients often experience severe complications, including chronic kidney disease, hypertension, neurological deficits, and cardiovascular events [9,10]. The disease's unpredictable nature and potential for rapid progression to end-stage renal disease necessitate intensive medical interventions, including plasma exchange, dialysis, and, in some cases, kidney transplantation [11]. These treatments not only impose significant physical and emotional strain on patients and families but also contribute to considerable healthcare expenditures.
- Scope and Purpose
The primary aim of this document is to present recommendations to provide a standardized, evidence-based framework for early diagnosis, accurate differentiation, and timely management of aHUS. By consolidating current international recommendations with region-specific clinical realities, these guidelines seek to improve diagnostic precision, reduce delays in initiating appropriate therapy, and ultimately enhance patient outcomes. They are intended to support clinicians across specialties in recognizing aHUS as a distinct clinical entity, ensuring prompt referral, and guiding therapeutic decisions, particularly in relation to complement inhibition strategies.
- Target Audience
These guidelines are intended for a multidisciplinary audience of healthcare professionals involved in the diagnosis and management of thrombotic microangiopathies, with a primary focus on aHUS. This includes pediatricians, internists, and nephrologists
- Methodology
A comprehensive search for guidelines was undertaken to identify the most relevant guidelines to consider for adaptation.
Inclusion/ exclusion criteria followed in the search and retrieval of guidelines to be adapted:
• Selecting only evidence-based guidelines (guideline must include a report on systematic literature searches and explicit links between individual recommendations and their supporting evidence)
• Selecting only national and/or international guidelines
• Specific range of dates for publication (using Guidelines published or updated in 2015 and later)
• Selecting peer reviewed publications only
• Selecting guidelines written in English language
• Excluding guidelines written by a single author, not on behalf of an organization to be valid and comprehensive, a guideline ideally requires multidisciplinary input
• Excluding guidelines published without references as the panel needs to know whether a thorough literature review was conducted and whether current evidence was used in the preparation of the recommendations
All retrieved guidelines and consensus statements relevant to atypical Hemolytic Uremic Syndrome (aHUS) were screened and appraised using the AGREE II instrument (www.agreetrust.org) by at least three independent members of the Guideline Development Group (GDG). A threshold was set whereby only guidelines scoring above 50% in the ‘rigour of development’ domain were retained. Based on this appraisal, the GDG decided to adapt the recommendations derived from the KDIGO Controversies Conference on aHUS and C3 Glomerulopathy [12] and the role of complement in kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference [13], along with other high-quality international sources, to formulate context-specific guidance for the diagnosis and management of aHUS
Evidence assessment
According to WHO Handbook for Guidelines, we used the GRADE (Grading of Recommendations, Assessment, Development and Evaluation) approach to assess the quality of a body of evidence, develop and report recommendations. GRADE methods are used by WHO because these represent internationally agreed standards for making transparent recommendations. Detailed GRADE information is available on the following sites:
• GRADE working group: http://www.gradeworkingroup.org
• GRADE online training modules: http://cebgrade.mcmaster.ca/
• GRADE profile software: http://ims.cochrane.org/revman/gradepro
Table 2: Quality and Significance of the four levels of evidence in GRADE:

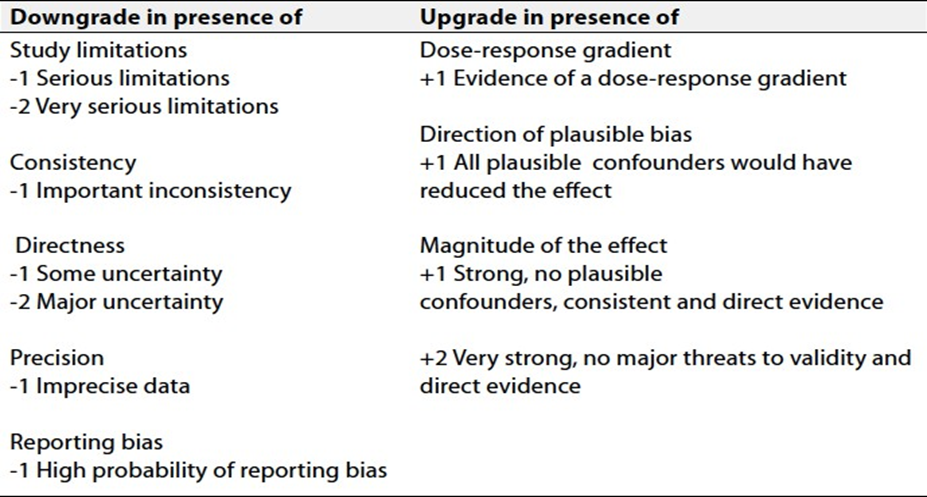
Table 3: Factors that determine how to upgrade or downgrade the quality of evidence
The strength of the recommendation
The strength of a recommendation communicates the importance of adherence to the recommendation.
With strong recommendations, the guideline communicates the message that the desirable effects of adherence to the recommendation outweigh the undesirable effects. This means that in most situations the recommendation can be adopted as policy.
These are made when there is greater uncertainty about the four factors above or if local adaptation has to account for a greater variety in values and preferences, or when resource use makes the intervention suitable for some, but not for other locations. This means that there is a need for substantial debate and involvement of stakeholders before this recommendation can be adopted as policy.
When not to make recommendations
When there is lack of evidence on the effectiveness of an intervention, it may be appropriate not to make a recommendation.
- Clinical Indicators for monitoring
· Urine output chart
· Blood pressure chart.
· Complete Blood Count (CBC).
· Kidney function, liver enzymes, electrolytes and blood gases.
· Coagulation profile
· Complete urine analysis and Pelvic-abdominal ultrasound.
· ESR, CRP, LDH, and serum ferritin.
· C3& C4.
· Retics, blood film, ADAMTS13 and H-factor if available.
· Stool analysis, Shiga-toxin if needed
- Research Gaps
1. Optimal Duration of Complement Inhibitor Therapy: No consensus on how long to continue complement inhibitors (Anti-C5 therapy).
2. Long-Term Safety and Outcomes of Complement Inhibitors: Limited long-term safety data (especially in children) on prolonged complement blockade.
3. Relapse Prediction and Early Detection: limitation of the presence of reliable biomarkers for relapse post-treatment.
4. The validity of liver transplantation in correcting complement dysregulation in aHUS remains unclear and requires further investigation.
- Update of guidelines
This guideline will be updated whenever there is new evidence
- References
1. Zuber J, Fakhouri F, Roumenina LT, Loirat C, Frémeaux-Bacchi V. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8:643–57.
2. Masias C, Vasu S, Cataland SR. None of the above: Thrombotic microangiopathy beyond TTP and HUS. Blood. 2017;129:2857–63.
3. Amirlak I, Amirlak B. Haemolytic uraemic syndrome: An overview. Nephrology. 2006;11:213–8.
4. Meri S. Complement activation in diseases presenting with thrombotic microangiopathy. Eur J Intern Med [Internet]. European Federation of Internal Medicine.; 2013;24:496–502. Available from: http://dx.doi.org/10.1016/j.ejim.2013.05.009
5. Wong EKS, Kavanagh D. Diseases of complement dysregulation — an overview . Seminars in Immunopathology 2018 DOI : https://doi.org/10.1007/s00281-017-0663-8 Date deposited : Seminars in Immunopathology; 2018;49–64.
6. Spasiano A, Palazzetti D, Dimartino L, Bruno F, Baccaro R, Pesce F, et al. Underlying Genetics of aHUS: Which Connection with Outcome and Treatment Discontinuation? Int J Mol Sci. 2023;24.
7. Juliette Leon, Marie-Bénédicte LeStang RS-S, Aude Servais, Dany Anglicheau VF-B, Zuber J. Complement‐driven hemolytic uremic syndrome. Am Jounral oh Hematol. 2023;98:S44–56.
8. Registry B, Uremic AH. B razilian S ociety of N ephrology Atypical Hemolytic Uremic Syndrome ( aHUS ): an expert consensus statement from the Rare Diseases Committee. 2025;1–22.
9. Bogdan RG, Anderco P, Ichim C, Cimpean AM, Todor SB, Glaja-Iliescu M, et al. Atypical Hemolytic Uremic Syndrome: A Review of Complement Dysregulation, Genetic Susceptibility and Multiorgan Involvement. J Clin Med. 2025;14:1–20.
10. Halimi JM, Al-Dakkak I, Anokhina K, Ardissino G, Licht C, Lim WH, et al. Clinical characteristics and outcomes of a patient population with atypical hemolytic uremic syndrome and malignant hypertension: analysis from the Global aHUS registry. J Nephrol [Internet]. Springer International Publishing; 2023;36:817–28. Available from: https://doi.org/10.1007/s40620-022-01465-z
11. Zuber, Julien, Moglie Le Quintrec, Heather Morris, Véronique Frémeaux-Bacchi, Chantal Loirat CL. Targeted strategies in the prevention and management of atypical HUS recurrence after kidney transplantation. Transplant Rev. 2013;27:117–25.
12. Goodship THJ, Cook HT, Fakhouri F, Fervenza FC, Frémeaux-Bacchi V, Kavanagh D, et al. Atypical hemolytic uremic syndrome and C3 glomerulopathy: conclusions from a “Kidney Disease: Improving Global Outcomes” (KDIGO) Controversies Conference. Kidney Int. 2017;91:539–51.
13. Vivarelli M, Barratt J, Beck LH, Fakhouri F, Gale DP, Goicoechea de Jorge E, et al. The role of complement in kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2024;106:369–91.
14. Laurence J. Atypical hemolytic uremic syndrome (aHUS): making the diagnosis. Clin Adv Hematol Oncol. 2012;10:1–12.
15. Noris M, Remuzzi G. Atypical Hemolytic–Uremic Syndrome. N Engl J Med. 2009;361:1676–87.
16. Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal Complement Inhibitor Eculizumab in Atypical Hemolytic–Uremic Syndrome. N Engl J Med. 2013;368:2169–81.
17. Laurence J. Atypical hemolytic uremic syndrome (aHUS): treating the patient. Clin Adv Hematol Oncol. 2013;11:4–15.
18. Frémeaux-Bacchi CL and V. Atypical hemolytic uremic syndrome. Orphanet J Rare Dis Rare Dis. 2011;6:1–30.
19. Zini G, d’Onofrio G, Briggs C, Erber W, Jou JM, Lee SH, et al. ICSH recommendations for identification, diagnostic value, and quantitation of schistocytes. Int J Lab Hematol. 2012;34:107–16.
20. Schapkaitz E, Mezgebe MH. The Clinical Significance of Schistocytes: A Prospective Evaluation of the International Council for Standardization in Hematology Schistocyte Guidelines. Turkish J Hematol. 2017;34:59–63.
21. Noutsos T, Laidman AY, Survela L, Arvanitis D, Segalla R, Brown SG, et al. An evaluation of existing manual blood film schistocyte quantitation guidelines and a new proposed method. Pathology. 2021;53:746–52.
22. McFarlane PA, Bitzan M, Broome C, Baran D, Garland J, Girard LP, et al. Making the Correct Diagnosis in Thrombotic Microangiopathy: A Narrative Review. Can J Kidney Heal Dis. 2021;8.
23. Arif Asif , Tushar Vachharajani LS and AN. A Simplified Approach to the Diagnosis of Atypical HUS: Clinical Considerations and Practical Implications. Open Urol Nephrol J. 2014;7:91–100.
24. Kato H, Nangaku M, Hataya H, Sawai T, Ashida A, Fujimaru R, et al. Clinical guides for atypical hemolytic uremic syndrome in Japan. Clin Exp Nephrol. Springer Japan; 2016;20:536–43.
25. Sawai T, Nangaku M, Ashida A, Fujimaru R, Hataya H, Hidaka Y, et al. Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the joint committee of the Japanese society of nephrology and the Japan pediatric society. Clin Exp Nephrol. 2014;18:4–9.
26. Cheong H Il, Jo SK, Yoon SS, Cho H, Kim JS, Kim YO, et al. Clinical practice guidelines for the management of atypical hemolytic uremic syndrome in Korea. J Korean Med Sci. 2016;31:1516–28.
27. Marie-Agnès Dragon-Durey, Sidharth Kumar Sethi, Arvind Bagga, Caroline Blanc, Jacques Blouin, Bruno Ranchin, Jean-Luc André, Nobuaki Takagi, Hae Il Cheong, Pankaj Hari, Moglie Le Quintrec, Patrick Niaudet, Chantal Loirat, Wolf Herman Fridman VF-B. Clinical features of anti-factor H autoantibody-associated hemolytic uremic syndrome. J Am Soc Nephrol. 2010;21:2180–7.
28. Józsi M, Licht C, Strobel S, Zipfel SLH, Richter H, Heinen S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. 2008;111:1512–4.
29. Shereen Shawky, Hesham Safouh, Mona Gamal, Mohammed M. Abbas, Azza Aboul-Enein, Toshihiro Sawai, Yosra Fahmy and HS. Anti‐Factor H Antibodies in Egyptian Children with Hemolytic Uremic. Int J Nephrol. 2021;6904858.
30. Johannes Hofer , Andreas R Janecke, L B Zimmerhackl, Magdalena Riedl, Alejandra Rosales, Thomas Giner, Gerard Cortina, Carola J Haindl, Barbara Petzelberger, Miriam Pawlik, VerenaJohannes Hofer 1, Andreas R Janecke, L B Zimmerhackl, Magdalena Riedl, Aleja RW. Complement factor H-related protein 1 deficiency and factor H antibodies in pediatric patients with atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2013;8:407–15.
31. Schaefer F, Ardissino G, Ariceta G, Fakhouri F, Scully M, Isbel N, et al. Clinical and genetic predictors of atypical hemolytic uremic syndrome phenotype and outcome. Kidney Int. 2018;94:408–18.
32. Khandelwal P, Bagga A. Managing anti-factor H antibody-associated hemolytic uremic syndrome: time for consensus. Pediatr Nephrol [Internet]. Springer Berlin Heidelberg; 2024;39:3137–41. Available from: https://doi.org/10.1007/s00467-024-06374-w
33. Fengxiao Bu , Tara Maga, Nicole C Meyer, Kai Wang, Christie P Thomas, Carla M Nester RJHS. Comprehensive genetic analysis of complement and coagulation genes in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2014;25:55–64.
34. Marina Noris , Jessica Caprioli , Elena Bresin , Chiara Mossali , Gaia Pianetti , Sara Gamba , Erica Daina , Chiara Fenili , Federica Castelletti , Annalisa Sorosina , Rossella Piras , Roberta Donadelli , Ramona Maranta, Irene van der Meer, Edward M Conwa GR. Relative Role of Genetic Complement Abnormalities in Sporadic and Familial aHUS and Their Impact on Clinical Phenotype. Clin J Am Soc Nephrol. 2010;5:1844–59.
35. Cataland SR, Wu HM. How I treat: The clinical differentiation and initial treatment of adult patients with atypical hemolytic uremic syndrome. Blood. 2014;123:2478–84.
36. Murphy D, Jha V, Banerjee D. Diabetes and CKD. Manag Kidney Dis. 2023;105:147–66.
37. Kurihara S, Yamaguchi A, Sonoda K, Yamada Y, Harada M, Hashimoto K, et al. Anti-C5 monoclonal antibody treatment showing pathological resolution of complement-mediated atypical hemolytic uremic syndrome: a case report. BMC Nephrol [Internet]. BioMed Central; 2024;25:1–10. Available from: https://doi.org/10.1186/s12882-024-03662-3
38. Sinha A, Gulati A, Saini S, Blanc C, Gupta A, Gurjar BS, et al. Prompt plasma exchanges and immunosuppressive treatment improves the outcomes of anti-factor H autoantibody-associated hemolytic uremic syndrome in children. Kidney Int. 2014;85:1151–60.
39. Boyer O, Balzamo E, Charbit M, Biebuyck-Gougé N, Salomon R, Dragon-Durey M-A, et al. Pulse Cyclophosphamide Therapy and Clinical Remission in Atypical Hemolytic Uremic Syndrome With Anti–Complement Factor H Autoantibodies. Am J Kidney Dis. 2010;55:923–7.
40. Kwon T, Dragon-Durey MA, Macher MA, Baudouin V, Maisin A, Peuchmaur M, et al. Successful pre-transplant management of a patient with anti-factor H autoantibodies-associated haemolytic uraemic syndrome. Nephrol Dial Transplant. 2008;23:2088–90.
41. Le Quintrec M, Zuber J, Noel LH, Thervet E, Frémeaux-Bacchi V, Fridman WH, et al. Anti-factor H autoantibodies in a fifth renal transplant recipient with atypical hemolytic and uremic syndrome. Am J Transplant. 2009;9:1223–9.
42. Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program. 2012;2012:617–25.
43. Loirat C, Fakhouri F, Ariceta G, Besbas N, Bitzan M, Bjerre A, et al. An international consensus approach to the management of atypical hemolytic uremic syndrome in children. Pediatr Nephrol. 2016;31:15–39.
44. Fadi Fakhouri, Marc Fila, François Provôt, Yahsou Delmas, Christelle Barbet, Valérie Châtelet, Cédric Rafat, Mathilde Cailliez, Julien Hogan, Aude Servais, Alexandre Karras, Raifah Makdassi, Fadi Fakhouri 1, Marc Fila, François Provôt, Yahsou Delmas, Chri VF-B. Pathogenic Variants in Complement Genes and Risk of Atypical Hemolytic Uremic Syndrome Relapse after Eculizumab Discontinuation. Clin J Am Soc Nephrol. 2017;12:50–9.
45. Maritati F, Corradetti V, Bini C, Provenzano M, Cuna V, Busutti M, et al. “Eculizumab First” in the Management of Posttransplant Thrombotic Microangiopathy. Kidney Int Reports [Internet]. Elsevier Inc; 2024;9:982–93. Available from: https://doi.org/10.1016/j.ekir.2024.01.013
46. Andrew M. Siedlecki, Nicole Isbel, Johan Vande Walle JJE, Cohen DJ. Eculizumab Use for Kidney Transplantation in Patients With a Diagnosis of Atypical Hemolytic Uremic Syndrome. Kidney Int Reports. 2019;4:434–46.
47. Aydan Mütiş Alan, Mevlüt Tamer Dinçer, Ata Kaykioğlu, Bahar Türk, Eda Nuhoğlu Kantarci, Nurhan Seyahi, Sinan Trabulus AEE. Perioperative Management of Atypical Hemolytic Uremic Syndrome in a Patient on Maintenance Eculizumab Therapy: A Case Report and Review of the Literature. Nephron. 2025;40414204.
48. Anuja Java. Peri- and Post-operative Evaluation and Management of Atypical Hemolytic Uremic Syndrome (aHUS) in Kidney Transplantation. Adv Chronic Kidney Dis. 2020;27:128–37.
49. Ha Tran, Abanti Chaudhuri WC& PCG. Use of eculizumab and plasma exchange in successful combined liver–kidney transplantation in a case of atypical HUS associated with complement factor H mutation. Pediatr Nephrol. 2014;29:477–80.