Diagnosis and management of Guillian Barre Syndrome in children and adolescents
| Site: | EHC | Egyptian Health Council |
| Course: | Pediatrics Guidelines |
| Book: | Diagnosis and management of Guillian Barre Syndrome in children and adolescents |
| Printed by: | Guest user |
| Date: | Thursday, 6 August 2026, 7:01 AM |
Description
"last update: 3 Nov 2025" Download Guideline
- Executive summary
1- The diagnosis of GBS should be based on clinical characteristics, a relevant constellation of clinical findings, and the exclusion of other relevant potential causes.
Requirements for the diagnosis of sensory- motor or motor GBS are:
• Bilateral flaccid paralysis of extremities
• Absent or decreased deep tendon reflexes in affected limbs
• Progressive worsening for no more than 4 weeks (strong recommendation)
2- Diagnostic lumbar puncture with subsequent CSF protein analysis and a cell count is done when the diagnosis is uncertain or when an alternative diagnosis needs to be excluded. Results supportive of GBS are an increased CSF protein concentration, and a normal or only slightly increased CSF white blood cell count (usually <5 cells/μL, rarely 5–50 cells/μL (known as cyto- albuminological dissociation) . A CSF white blood cell count of >50 cells/μL should raise suspicion for alternative diagnoses (strong recommendation)
We suggest that for diagnostic certainty in the event of an initially-normal CSF finding, a lumbar puncture may be repeated 7-10 days after the first symptoms appear, if it is deemed necessary due to unclear clinical or electrophysiological results. (Good practice statement)
3- Electrophysiological examination for confirming GBS diagnosis is recommended as it is necessary for obtaining the highest level of diagnostic certainty ( Strong recommendation). If the diagnostic tool is not available, refer the patient to an appropriate clinical centre.
We suggest that for confirmation of diagnosis in the event of an initially normal electrophysiological finding, the procedure may be repeated 1 or 2 weeks later if it is deemed necessary because of other unclear data. (conditional recommendation)
4- Performing MRI of brain and spinal cord is done when the available findings are ambiguous or if there is evidence of pathology at either the spinal level (e.g., bladder dysfunction at the onset of disease, definable motor or sensory lesion) or central level (encephalopathy, pyramidal signs) (strong recommendation)
5- Microbiological and serological diagnostics may be carried out as the diagnosis of GBS is more likely if there is a history of recent infection. Asking about these antecedent events may increase diagnostic confidence and is generally more useful when present than absent (Conditional recommendation)
6- It is not advised to do routine testing for serum antibodies against gangliosides in most patients with motor-sensory GBS, because of their low–moderate sensitivity and frequent delay in reporting results of antibody assays beyond the therapeutic window (good practice statement)
If MFS (or MFS spectrum) is suspected, test for serum antibodies against GQ1b can be done especially when there is some clinical doubt and test results can be obtained within reasonable time (Conditional Recommendation)
7- Starting IVIg or plasma exchange as soon as possible is recommended in patients unable to walk unaided (GBS- DS grade 3 or more) if still within the first 2 weeks from onset of weakness ( strong recommendation)
• Using the most frequently used and proven effective standard course of IVIg (0.4 g/kg/day for 5 days) rather than a low- dose (0.4 g/kg/day for 3 days) or a high- dose (0.4 g/kg/day for 6 days) regimen or a 2- day regimen (1 g/kg/day) or using four to five plasma exchanges over 1– 2 weeks with a total exchanged volume of 12– 15 L (good practice statement)
We advise to start IVIg (or PE four to five exchanges) in patients who are still able to walk unaided (GBS- DS grade 2) within 4 weeks from onset of weakness, but who have a fast rate of deterioration, a risk of requiring ventilatory support, swallowing difficulties, autonomic disturbances or poor prognostic factors (good practice statement)
9- We don’t recommend the use of oral corticosteroids or IV methylprdnisone (IVMP) for the treatment of GBS. (strong recommendation)
10- We don’t recommend using eculizumab for treatment of GBS.(conditional recommendation) because of the lack of demonstrated efficacy, the known adverse effects (all patients had some adverse effects) and the high cost currently.
11- We advise using a prognostic model at hospital admission to quantify the risk of requiring mechanical ventilation. This can be quantified using the modified Erasmus GBS Respiratory Insufficiency Score (mEGRIS)( table4) OR
Using the following risk factors for requiring mechanical ventilation during hospital admission:
Rapid progression of limb weakness during hospital admission
GBS- DS grade 4 (unable to walk 5 m even with aid)
Neck flexion, facial, or bulbar weakness
The inability to cough
Autonomic instability such as fluctuations in blood pressure or heart rate. (good practice statement)
• We advise regularly assessing any decline in respiratory function by measuring forced vital capacity (FVC), and single breath count (SBC). Fig (good practice statement)
12- Changing the diagnosis from GBS to A-CIDP is suggested after a few weeks from onset in some patients initially diagnosed with GBS, especially if the patient worsens after initial improvement or stabilization (known as a treatment-related fluctuation, TRF), or presents as mild or slowly progressive GBS and continues to worsen.(conditional recommendation)
A-CIDP is more likely if there are three (or more) TRFs.
A-CIDP cannot be confirmed unless there is further worsening at least 8 weeks after onset.
In case of a TRF, consider re-treatment with IVIg or PE.(good practice statement)
- Recommendations
PICO 1: What are the diagnostic criteria of GBS?
Requirements for the diagnosis of sensory- motor or motor GBS are:
• Bilateral flaccid paralysis of extremities
• Absent or decreased deep tendon reflexes in affected limbs
• Progressive worsening for no more than 4 weeks (strong recommendation, high grade evidence)
Evidence Summary:
Features that support diagnosis:
1-Relative symmetry, 2-Relatively mild/absent sensory symptoms and signs,3- Cranial nerve involvement (especially bilateral facial palsy), 4- Autonomic dysfunction, 5- Respiratory insufficiency (due to muscle weakness), 6- Pain (muscular/radicular in back or limb),7- Recent history of infection (<6 weeks).(16)
To ensure the highest level of diagnostic certainty, additional proof is required in the form of increased protein levels in the CSF, along with a mildly increased cell count, and electrophysiological signs of neuropathy.
The maximal duration of progression originates from large studies showing that progression does not exceed 2 weeks in most patients, and almost never exceeds 4 weeks.(9,10)
2- Cerebrospinal fluid analysis:
PICO2: Cerebrospinal fluid (CSF) examination: In patients with clinically suspected GBS, does examination of the CSF compared with no CSF examination influence the diagnostic accuracy, treatment response and patient outcome?
1- We strongly recommend a diagnostic lumbar puncture with subsequent CSF protein analysis and a cell count particularly when the diagnosis is uncertain or when an alternative diagnosis needs to be excluded (strong recommendation, moderate grade evidence)
2- Results supportive of GBS are an increased CSF protein concentration, and a normal or only slightly increased CSF white blood cell count usually <5 cells/μL, rarely 5–50 cells/μL (known as cyto- albuminous dissociation) (Strong recommendation, moderate grade evidence)
3- A CSF white blood cell count of >50 cells/μL should raise suspicion for alternative diagnoses (Strong recommendation, moderate grade evidence)
4- We suggest that for diagnostic certainty in the event of an initially-normal CSF finding, a lumbar puncture may be repeated 7-10 days after the first symptoms appear, if it is deemed necessary due to unclear clinical or electrophysiological results .(Good practice statement)
Evidence summary:
- Normal CSF protein does not exclude GBS. It should be noted that protein concentration is often normal during the first week, and only increases with disease progression so, diagnostic sensitivity of an increased CSF protein depends on the time CSF is examined after onset of weakness. After 7-10 days, it generally reaches a level above that of the reference value, which notably differs in children according to age [11].
- The specificity of a raised CSF protein is unknown, and it does not rule out some mimics of GBS as CSF protein levels can be elevated in other disorders that are similar to GBS.. Therefore, a normal CSF protein reading does not rule out GBS, while an elevated protein level is not solely sufficient to confirm diagnosis. [12]
- Nevertheless, the CSF cell count is usually normal, although occasionally it can be mildly elevated to <50/mm3, rarely more [13]. In such cases, other differential diagnoses as meningitis should be considered as possible causes [13]. Due to the fact that the GBS-related increase in protein is dependent on time, and the standard CSF value for protein depends on age, the cell count can potentially be more valuable as a differential diagnostic factor than protein level.
- Both CSF protein and cell count may be artefactually raised after IVIG [14]
- Since this is an invasive procedure that can cause discomfort and be stressful for children and often has no therapeutic consequences, it is preferred not to be repeated except if unclear clinical or electrophysiological results. [15]
3- Electrophysiological examination:
PICO 3: In patients with clinically suspected GBS, Regarding the added value of electrophysiological examination?
1- We recommend performing electrophysiological examination for confirming GBS diagnosis as it is necessary for obtaining the highest level of diagnostic certainty (strong recommendation, low grade evidence)
If the diagnostic tool is not available, refer the patient to an appropriate clinical centre..
2- We suggest that for confirmation of diagnosis in the event of an initially normal electrophysiological finding, the procedure may be repeated 1 or 2 weeks later if it is deemed necessary because of other unclear data. (conditional recommendation, moderate grade evidence)
Evidence summary:
Prospective and retrospective studies that evaluated up to 84 patients with clinically suspected GBS or up to 66 AIDP cases with a variable number of controls concluded that numerous nerve conduction parameters are abnormal early after disease onset.(19,20) However, some studies included small numbers of patients and/or did not include controls. Sensitivity and specificity vary according to electrodiagnostic criteria and the control groups used.
A typical electrodiagnostic finding is required in order to reach the highest level of diagnostic certainty described by Sejvar et al. [18] Such examinations can be painful and are often difficult to perform in children. Moreover, the necessary expertise is often lacking in pediatric hospitals. Nonetheless, nerve conduction studies and, to a lesser extent, EMG, are required for distinguishing between GBS variants. Notably, due to the latent appearance of pathological spontaneous activity it only makes sense to perform an EMG 2-3 weeks after onset.
Although electrophysiological criteria for the diagnosis of GBS and its variants have been repeatedly defined, they are not always consistently applied and cannot be considered definitive [21,22].
4- MRI brain and spine
PICO (4) In patients with clinically suspected GBS, does using MRI brain and/or spine compared with not using MRI studies influence the diagnostic accuracy .
We recommend performing MRI of the brain and spinal cord when the available findings are ambiguous or if there is evidence of pathology at either the spinal level (e.g., bladder dysfunction at the onset of disease, definable motor or sensory lesion) or central level (encephalopathy, pyramidal signs). (strong recommendation, low grade evidence)
Evidence summary:
Although a spinal MRI is primarily carried out to exclude space-occupying lesions or any other pathology, a contrast enhanced MRI procedure could also support the diagnosis of GBS. A number of publications has reported pathological uptake of contrast agent, particularly by the ventral nerve roots and cranial nerves, hence suggesting that a contrast-enhanced MRI could serve as a diagnostic criterium for GBS in the face of persistently unremarkable CSF and nerve conduction findings.(23,24)
In a retrospective study by Yikilmaz et al. 38 of 40 patients with childhood GBS showed contrast enhancement of the nerve roots; Mulkey et al. reported 22 of 24 patients with spinal nerve root enhancement in an initial MRI(25,26). However, increased uptake of contrast agent is also possible in other inflammatory conditions (serous meningitis, neuroborreliosis) and neurometabolic disorders (Krabbe disease). Since prospective examinations have not yet been carried out, the sensitivity and specificity of contrast-enhanced MRI remains unclear in children and adolescents with GBS.
5- Antecedent events :
PICO (5) In patients with clinically suspected GBS, does enquiring about antecedent events ( infection or vaccination) compared with not enquiring about antecedent events influence the diagnostic test accuracy, treatment response and patient outcome?
Infection:
We suggest that microbiological and serological diagnostics may be carried out as the diagnosis of GBS is more likely if there is a history of recent infection. Asking about these antecedent events may increase diagnostic confidence and is generally more useful when present than absent (Conditional recommendation, moderate grade evidence)
Evidence Summary:
About two- thirds of patients who develop GBS report symptoms of an infection in the 6 weeks preceding the onset of the condition (27) These infections are thought to trigger the immune response that causes GBS. In childhood GBS, a preceding infection is identified as the potential causative agent in 60-70% of cases.
Six pathogens have been temporally associated with GBS in case–control studies: Campylobacter jejuni, cytomegalovirus, hepatitis E virus, Mycoplasma pneumoniae, Epstein–Barr virus and Zika virus.(28,29) It has been suggested that other pathogens are linked to GBS on the basis of evidence from case series or epidemiological studies, but their role in the pathogenesis of GBS is uncertain.
GBS is not or marginally increased in incidence after SARS- CoV- 2 infections although large case– control studies are lacking, from the epidemiological and cohort studies that exist.(30,31)
In general, the absence of an antecedent illness does not exclude a diagnosis of GBS, as putative infections or other immunological stimuli can be subclinical.
Vaccination :
We recommend thorough microbiological and serological diagnostic tests if GBS occurs within 1-6 weeks of vaccination for investigating a potential link with the vaccinating antigen, as well as with alternative causal agents. (Strong recommendation, moderate grade evidence)
Evidence Summary:
The results arising from microbiological and serological diagnostic tests can have substantial legal implications at the social and health care levels.
GBS has been associated temporally with numerous vaccines; however, such temporal association must be differentiated from causality. In general, specific biological markers indicative of a cause-and-effect association with a particular pathogen or vaccine are absent.(32)
Well-conducted case-control studies have been able to show that most vaccines have a very unlikely causal link to GBS. The only exception comes from a swine flu vaccine that was used in an American immunization campaign in 1976-77 and led to a significant increase in the number of GBS cases. This effect was not observed again in later immunization campaigns, even after vaccination during the recent swine flu pandemic in 2009-10.(33)
Furthermore, an accumulation of GBS cases was observed 30-40 years ago following immunization with a rabies vaccine that was grown on mammalian brain tissue. This, however, has not been observed in association with modern rabies vaccines.(34)
6-Serum antibodies
PICO (6) In patients with clinically suspected GBS, does testing for serum antibodies compared with not testing for these antibodies influence the diagnostic test accuracy, particularly for GBS subtypes such as AMAN and Miller Fisher syndrome?
We don’t advise to do routine testing for serum antibodies against gangliosides in most patients with motor-sensory GBS, because of their low–moderate sensitivity and frequent delay in reporting results of antibody assays beyond the therapeutic window (good practice statement)
We suggest testing for serum antibodies against GQ1b in patients suspected to have MFS (or MFS spectrum) especially, when there is some clinical doubt and test results can be obtained within reasonable time (Conditional Recommendation, moderate grade evidence )
Summary of evidence:
Serum antibodies that have been tested in GBS cases include GM1, GM1b, GD1a, GQ1b, GalNAc- GD1a, GT1a, GD1b, GD3, O-GD3, GT3, O-GT3, sulfatide, galactocerebroside, CNTN1, NF155 and NF186, cardiolipin, LM1, sulphated glycolipids, P0 and PMP22. The test accuracy varies depending on GBS subtype, tested antigen and control group. (35,36)
In GBS, the sensitivity of anti-ganglioside antibody panels is reported to vary between 32% and 64%, depending on the presence of a recent infection or GBS subtype (AIDP or AMAN).(37)
For MFS, sensitivity for anti-GQ1b antibodies is high (88%–100%), with a very high specificity (100%). Especially, when there is some clinical doubt and test results can be obtained within reasonable time, testing for anti-GQ1b antibodies is considered helpful. (38,39)
1- Therapeutic recommendations:
PICO (7) In patients with clinically suspected GBS ,Is treatment with intravenous immunoglobulins (IVIG) recommended in children with GBS?
We recommend starting IVIg as soon as possible in patients unable to walk unaided (GBS- DS grade 3 or more) if still within the first 2 weeks from onset of weakness ( strong recommendation- high level of evidence)
• Using the most frequently used and proven effective standard course of IVIg (0.4 g/kg/day for 5 days) rather than a low- dose (0.4 g/kg/day for 3 days) or a high- dose (0.4 g/kg/day for 6 days) regimen or a 2- day regimen (1 g/kg/day) (good practice statement)
We advise to start IVIg (or PE) also in patients who are still able to walk unaided (GBS- DS grade 2) within 4 weeks from onset of weakness, but who have a fast rate of deterioration, a risk of requiring ventilatory support, swallowing difficulties, autonomic disturbances or poor prognostic factors (good practice statement)
Summary of evidence :
In the absence of systematic dose-ranging study data, IVIG is usually administered in children as it is in adults with acute GBS, namely with a single cycle of 2 g/kg body weight distributed over 4 - 5 consecutive days. Reducing the period of IVIG administration to 2 g/kg in 2 days did not show any improved effects but was associated with a higher frequency of relapses [40].
The most common side-effects were headache (12.8% of infusions), fatigue (5.2%), stomach-ache (2.3%) and myalgia (2.3%). Acute side-effects such as skin rash, fever, bronchospasm and thoracic pain occurred more rarely (0.3-0.6%) [41].
An initially progressive disease course with a subsequent plateau phase without recovery is classified as severe GBS, for which the optimal treatment approach remains unclear . However, in children a satisfactory recovery can still be expected even after a prolonged plateau-phase of up to 90 days[42].
8-Plasmapheresis, plasma exchange
PICO ( 8) Is treatment with plasmapheresis recommended in children with GBS?
The same indications as listed in the IVIG in cases when IVIG is not available. We recommend four to five exchanges over 1– 2 weeks with a total exchanged volume of 12– 15 L in patients who are severely disabled (unable to walk unaided, bedridden or ventilated). (strong recommendation, high grade evidence)
We suggest PE (four to five exchanges over 1– 2 weeks) in patients who are still ambulatory, but who have a fast rate of deterioration, a risk of requiring ventilatory support, swallowing difficulties or other poor prognostic factors. These patients are considered at high risk of further deterioration, which may potentially be prevented by starting treatment early( good practice statement)
Summary of evidence
The best effect of PE is seen when therapy is started within seven days of symptom onset, although an effect can still be detected if applied up to 30 days post-onset.
The given dosages for PE mostly comprise 4-5 cycles with an exchange volume of 200-250 ml/kg body weight over 7-14 days.
Albumin is a more favorable exchange fluid than FFP [43].
There is no preference for treatment with either IVIg or PE. Therefore, the choice may depend on the availability or the experience.(50) However, IVIg is generally associated with few adverse events and is readily available in most hospitals. PE requires special facilities, good intravenous access and has a slightly higher adverse event (AE) rate [42].
The combination of PE followed by IVIG did not yield any significant additional benefits, although an effect in individual patients cannot be ruled out.
Application of PE within 1-2 weeks of the preceding IVIG therapy is not indicated.
PICO (9). In patients diagnosed with GBS, does treatment with corticosteroids (IV or oral), alone or in combination with IVIg or PE, influence the disease course ?
We don’t recommend the use of oral corticosteroids or IV methylprdnisone (IVMP) for the treatment of GBS.(strong recommendation, moderate level of evidence)
Summary of evidence:
There is a probable lack of efficacy of IVMP (moderate certainty evidence from a trial with 242 patients),(44)
The probable lack of efficacy of the combination of IVIg and IVMP compared with IVIg and placebo (moderate certainty evidence from a trial with 225 patients)(45)
The probable harm (delayed recovery) of oral corticosteroids (low certainty evidence from 4 trials with a total of 120 patients) using various oral regimens of the equivalent of 40 mg prednisolone daily for at least 2 weeks, and the high certainty evidence of adverse effects (diabetes more common), despite that hypertension was less common in the corticosteroid-treated patients, which is crucial in decision making. (46,47,48)
PICO (10 ). Does treatment with pharmacological therapies other than PE, IVIg (as eculizumab, mycophenolate, interferons or other) influence disease course ?
We don’t recommend the use of immunosuppressive treatment approaches as an alternative to immunomodulation with plasmapheresis and IVIG. (conditional recommendation, low grade evidence)
Summary of evidence:
As there is evidence for complement activation from pathological studies and from animal models of GBS.(53) Eculizumab, a complement blocking agent, was beneficial in an animal model.(54)
In two small phase 2 trials (including a total of 41 patients), no beneficial effects of eculizumab could be demonstrated (very low to low certainty evidence).(55)
PICO (11) In patients diagnosed with GBS, what is indication of ICU admission and mechanical ventillation?
We advise using a prognostic model at hospital admission to quantify the risk of requiring mechanical ventilation. This can be quantified using the modified Erasmus GBS Respiratory Insufficiency Score (mEGRIS)( table5) OR
Using the following risk factors for requiring mechanical ventilation during hospital admission. These include:
- Rapid progression of limb weakness during hospital admission
- GBS- DS grade 4 (unable to walk 5 m even with aid)
- Neck flexion, facial or bulbar weakness
- The inability to cough
- Autonomic instability such as fluctuations in blood pressure or heart rate. (good practice statement)
• We advise regularly assessing any decline in respiratory function by measuring forced vital capacity (FVC), and single breath count (SBC). (good practice statement) Fig 2
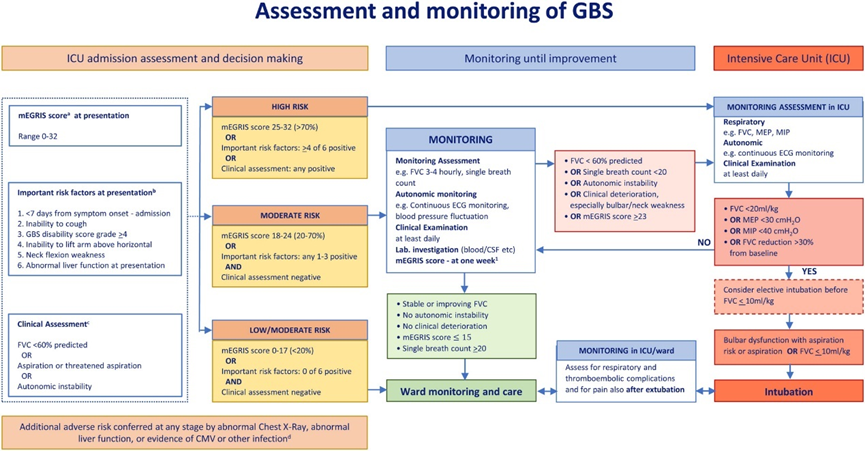
Fig.2: Assessment and monitoring of GBS
Frequent monitoring of the following quantitative measures of ventilatory function:
a- FVC should be checked between three and six times a day, depending on severity, and until significant worsening seems unlikely. While the patient is still declining and there is a reduction in FVC, 4- hourly monitoring is likely appropriate.
b- A fall of the FVC > 30% below the predicted baseline should alert concern, a fall of >30% in 24 h likely indicates immediate transfer to ICU, or a 50% decline in under 24 h likely indicates the need for ventilation.
c- Single breath count <20 (inability to count in a single breath out loud from 1 to 20 indicates the need to transfer to ICU.
Summary of evidence :
According to the published case series on GBS in childhood, 15-25% of patients were dependent on mechanical ventilation. Respiratory failure in children particularly needs to be reckoned with when the disease progresses quickly and the upper extremities and cranial nerves are affected (56,57) Moreover following discharge from the ICU the possibility of secondary deterioration in the ensuing weeks should be kept in mind.
PICO (12). In patients initially diagnosed with GBS, does the presence of certain clinical symptoms or laboratory features compared with their absence predict the subsequent diagnosis of A-CIDP as confirmed by neurological worsening at >8 weeks from onset?
We suggest changing the diagnosis from GBS to A-CIDP after a few weeks from onset in some patients initially diagnosed with GBS, especially if the patient worsens after initial improvement or stabilization (known as a treatment-related fluctuation, TRF), or presents as mild or slowly progressive GBS and continues to worsen.(conditional recommendation, low grade evidence)
• A-CIDP is more likely if there are three (or more) TRFs.
• A-CIDP cannot be confirmed unless there is further worsening at least 8 weeks after onset.
• In case of a TRF, consider re-treatment with IVIg or PE.(good practice statement)
Evidence summary:
In a prospective study of 170 patients initially diagnosed as GBS, 16 (9%) had a TRF and another 8 (5%) were subsequently diagnosed with A-CIDP (58). Confirmation of these results however warrants additional studies in larger numbers of patients.
A-CIDP is likely in patients initially diagnosed with GBS, if there is further worsening after 8 weeks from onset (sensitivity 100%, specificity 92%)(51,52), or when there are three (or more) TRFs (episodes of worsening following treatment-induced improvement/stabilisation) (sensitivity 52%, specificity 96%) (49,59).
Reduced MNCV < 90% of lower limit of normal (or <85% if small distal CMAP) was more frequently found in A-CIDP than in GBS . Patients with A-CIDP tested with nerve US within 4 weeks of onset had greater enlargement of peripheral nerves compared with those with GBS (sensitivity 88%, specificity 84%) (60).. Some patients suspected to have A-CIDP may have an autoimmune nodopathy (61). These patients have a poorer response to conventional therapies for CIDP, and there is anecdotal evidence that these patients may response to rituximab. There is no evidence from an RCT, but observational data indicate that a repeated course of IVIg or PE can be effective in case of a TRF(62)
Rationale: It is important to diagnose A-CIDP because treatment differs from GBS. A-CIDP however should not be overdiagnosed in severely weak patients with slow or no improvement. If in doubt, the presence of muscle wasting and denervation on electromyography indicates that GBS-related axonal degeneration is more likely than A-CIDP.
- Committee
Chair of Guideline Development Group (GDG):
Prof. Dr. Marian Yousry Fahmy , Faculty of medicine ,Cairo University
Members of GDG :
Prof. Dr. Hoda Yehia Tamoum,Faculty of medicine , Ain Shams university
Prof. Dr. Hanan Azouz , faculty of medicine , Alexandria university
Prof. Dr. Bahaa Elhawary ,Faculty of medicine , Asswan university,
Prof. Dr. Abdelreheem Abdrabo, Faculty of medicine , Sohag university
Prof. Dr. Samir Mounir , Faculty of medicine , Menia university
Prof . Dr. Eman Gad, Faculty of medicine , Assiut university
Prof. Dr. Walaa Elnagar, Faculty of medicine , Cairo university
Prof. Dr. Shimaa Maher, Faculty of medicine , Ain Shams university
Prof. Dr. Marwa Saeed , Faculty of medicine , Alexandria University
Prof. Dr. Heba ElAwady, Faculty of medicine , Fayoum university
Dr. Sherry Kodsy, Military Hospitals.
- Abbreviations
AE: Adverse events
AIDP :acute inflammatory demyelinating polyneuropathy
AMAN :acute motor axonal neuropathy .
AMSAN :acute motor and sensory axonal neuropathy .
BBE: Bickerstaff brainstem encephalitis
CIDP: Chronic inflammatory demyelinating polyneuropathy
CSF :cerebrospinal fluid
DS: Disability Scale
FFP: Fresh Frozen Plasma
FVC :forced vital capacity
GBS :Guillain Barre syndrome
ICU :intensive care unit.
IVIG :intravenous immunoglobulins
IVMP : IV methylprednisolone
mEGRIS :modified Erasmus GBS Respiratory Insufficiency Score
MFS :Miller Fisher Syndrome
MRC: Medical Research Council
PE :plasmapheresis
SBC: single breath count
TRF :treatment-related fluctuation,
- Introduction
Guillain Barre syndrome (GBS) is the most common cause of acute flaccid paralysis with an annual global incidence of approximately
1–2 per 100 000 person years(1,2) The diagnosis of GBS relies upon a combination of clinical features, often with support of electrodiagnostic and laboratory features. Most diagnostic criteria for GBS require a combination of history, neurological examination, cerebrospinal fluid (CSF)and electrodiagnostic results.(3)
GBS can be subclassified into a number of variants, depending on clinical presentation and electrophysiological findings (4) .The classic variants are acute demyelinating inflammatory polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN).
These are characterized by rapidly-progressing, ascending symmetrical weakness, with attenuation or loss of muscle proprioceptive reflexes. Among pediatric patients in the peak phase of the disease, 75% can no longer walk unaided, 30% are tetraparetic,35-50% show cranial nerve involvement, and 15-20% have respiratory failure and/or autonomic dysfunction. Furthermore, up to 70% suffer from neuropathic pain, which can be severe and occasionally occur as the first symptom. Localized variants of GBS include Miller Fisher Syndrome (MFS) (cranial nerve affection and ataxia, areflexia, serum anti-GQ1b antibody detection in >90% cases) and the pharyngeal-cervical-brachial variant (predominantly bulbar and neck weakness, serum IgG antibodies against GT1a frequently detected), both of which are extremely rare in childhood (5) .Immunomodulatory therapy with plasmapheresis (PE) or intravenous immunoglobulins (IVIG) has been proven effective in GBS namely by accelerating the improvement of weakness. (6,7)
The improvement follows a plateau phase that ranges from days to weeks and runs an extremely variable course, regardless of whether a specific therapy has been applied or not. The overall long-term prognosis for children with GBS is more favorable than that in adults, whereby the majority of children largely regain motor function.(8)
There are several forms of GBS within the disease spectrum, which are defined by the clinical features, with some overlap: fig 1
1- Motor-sensory((weakness with sensory symptoms and/or signs) and motor ((weakness without sensory symptoms or signs )GBS are considered ‘typical’ GBS
2- Pharyngeal–cervical–brachial GBS with weakness in the corresponding regions, bilateral facial weakness with limb paraesthesias (but no limb weakness)
3- Paraparetic variant with weakness starting in the legs that may evolve to tetraparesis
4- Pure sensory syndrome of numbness or tingling, reduced or absent reflexes, and sometimes ataxia or pseudoathetosis, but no weakness, with a time course like GBS. Nerve conduction in these patients may show signs of demyelination in sensory or motor nerves. Although the patients with this pure sensory syndrome do not fulfil all diagnostic criteria for GBS, this probably represents a rare sensory variant of GBS
5- MFS spectrum : Patients with typical MFS have ophthalmoplegia, ataxia and areflexia
Bickerstaff brainstem encephalitis (BBE) is considered a rare variant of MFS that clinically has a combination of ophthalmoplegia, ataxia, pyramidal tract signs and impaired consciousness.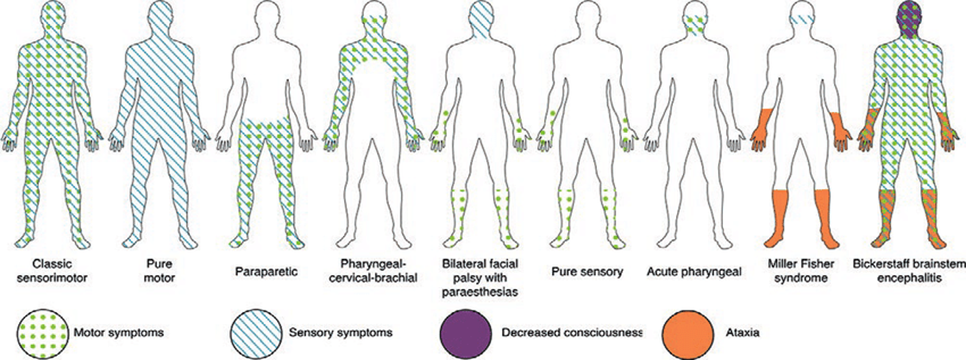
Fig 1: Forms of GBS
- Scope and purpose
The aim of this guideline is to provide practical recommendations for diagnostic procedures and the treatment of GBS in childhood and adolescence. The target population are children and adolescents presenting with clinical features suggestive of GBS. The aim is to optimize diagnostic accuracy and to improve the patient outcomes.
- Target audience
This guideline is intended for pediatricians, intensivists, pediatric neurologists and general practitioners who are involved in the care of pediatric population.
- Evidence assessment
According to WHO Handbook for Guidelines, we used the GRADE (Grading of Recommendations, Assessment, Development and Evaluation) approach to assess the quality of a body of evidence, develop and report recommendations. GRADE methods are used by WHO because these represent internationally agreed standards for making transparent recommendations. Detailed GRADE information is available on the following sites:
• GRADE working group: https://www.gradeworkinggroup.org/
• GRADE online training modules: http://cebgrade.mcmaster.ca/
Table 1 Quality and Significance of the four levels of evidence in GRADE:

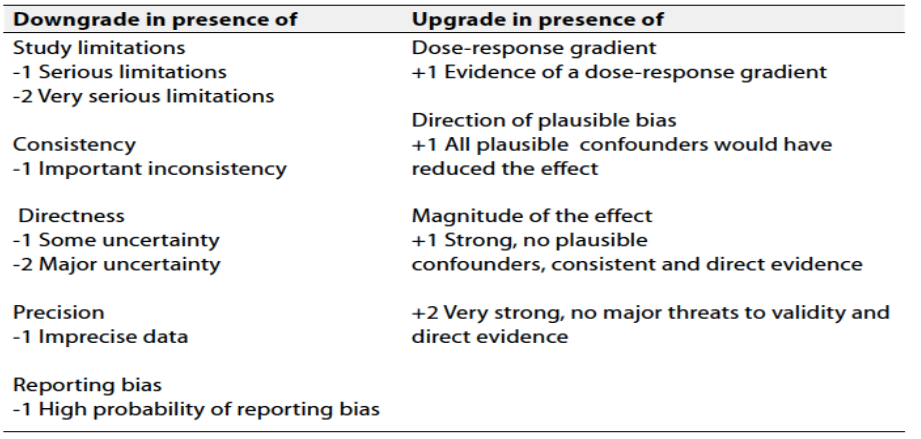
Table 2 Factors that determine How to upgrade or downgrade the quality of evidence
➡️The strength of the recommendation
The strength of a recommendation communicates the importance of adherence to the recommendation.
Strong recommendations
With strong recommendations, the guideline communicates the message that the desirable effects of adherence to the recommendation outweigh the undesirable effects. This means that in most situations the recommendation can be adopted as policy.
Conditional recommendations
These are made when there is greater uncertainty about the four factors above or if local adaptation has to account for a greater variety in values and preferences, or when resource use makes the intervention suitable for some, but not for other locations. This means that there is a need for substantial debate and involvement of stakeholders before this recommendation can be adopted as policy.
When not to make recommendations
When there is lack of evidence on the effectiveness of an intervention, it may be appropriate not to make a recommendation.
- Methods
This guideline has been developed by a group of dedicated pediatric neurology consultants volunteering from all over the Egyptian universities across Egypt. It is the result of an initiative held by the Egyptian Health Council, aiming to formulate evidence-based recommendations for clinical practice.
A comprehensive search for guidelines was undertaken to identify the most relevant guidelines to consider for adaptation.
Inclusion/ exclusion criteria followed in the search and retrieval of guidelines to be adapted:
• Selecting only evidence-based guidelines (guideline must include a report on systematic literature searches and explicit links between individual recommendations and their supporting evidence)
• Selecting only national and/or international guidelines
• Specific range of dates for publication (using Guidelines published or updated in 2015 and later)
• Selecting peer reviewed publications only
• Selecting guidelines written in English language
• Excluding guidelines written by a single author, not on behalf of an organization to be valid and comprehensive, a guideline ideally requires multidisciplinary input
• Excluding guidelines published without references as the panel needs to know whether a thorough literature review was conducted and whether current evidence was used in the preparation of the recommendations
The following characteristics of the retrieved guidelines were summarized in a table:
• Developing organization/authors
• Date of publication, posting, and release
• Country/language of publication
• Date of posting and/or release
• Dates of the search used by the source guideline developers
All retrieved Guidelines were screened and appraised using AGREE II instrument (www.agreetrust.org) by at least three members. The panel decided on a cut-off point or ranked the guidelines (any guideline scoring above 50% on the rigor dimension was retained).
The Guideline Development Group (GDG) decided to adapt the Guideline published by the German Speaking Society of Neuropediatrics (GNP) “Diagnosis and Treatment of Guillain Barre Syndrome in Childhood and adolescence: An evidence and Consensus Based guideline- 2020 AND the guideline published by the European Academy of Neurology/Peripheral Nerve Society on the Diagnosis and treatment of Guillain Barre syndrome- 2023.(16,17)
- Clinical indicators for monitoring
1-Documentation of symptoms and signs in the medical file:
Progressive weakness of arms and legs for no more than 4 weeks..
By examination: absent or decreased deep tendon reflexes in affected limbs.
2- Electrophysiological studies (showing evidence of neuropathy) if available.
3- CSF analysis (showing cyto-albuminuous dissociation), if diagnosis is uncertain
4- IVIg (0.4 g/kg/day for 5 days) or plasma exchange 4-5 sessions should be started if GBS-DS grade 3 or more in first 2 weeks OR grade 2 in patients who have a fast rate of deterioration, a risk of requiring ventilatory support, swallowing difficulties, autonomic disturbances or poor prognostic factors
5- Documentation of the risk factors for requiring mechanical ventilation
-Rapid progression of limb weakness during hospital admission
- GBS- DS grade 4 (unable to walk 5 m even with aid)
- Neck flexion, facial or bulbar weakness
- The inability to cough
- Autonomic instability such as fluctuations in blood pressure or heart rate.
6-Single breath count (SBC) to assess any decline in respiratory function.
- Research Gaps
Recommended further research in the field of GBS:
1. In Pathogenesis and Immune Mechanisms
- Better understanding of molecular mimicry between pathogens (e.g., Campylobacter jejuni) and gangliosides.
- Role of complement activation, T-cell subsets, and innate immunity in different GBS variants.
- Exploration of genetic susceptibility (e.g., HLA subtypes, complement-regulatory genes).
- Role of infections and vaccines.
2. Biomarkers and Early Diagnosis
- Development of CSF and blood biomarkers
- Neurofilament light chain (NfL) and anti-ganglioside antibodies as prognostic tools.
- Advanced imaging (e.g., nerve ultrasound, DTI MRI) to detect early inflammation or axonal injury.
3. Treatment options
Trials of novel immunotherapies, such as:
- Complement inhibitors (e.g., eculizumab)
- Anti-GM1 monoclonal antibodies
- T-cell modulation strategies
- Guideline update
This guideline will be updated whenever there is new evidence.
- Appendix
|
Table3 :GBS disability scale (Hughes and Cornblath) (45) |
|
0 - healthy 1 - minor symptoms or signs of neuropathy, but capable of manual work and running 2 - can walk without the aid of a stick for 5 m across an open space, but is not capable of manual work or running 3 - can walk with a stick, orthosis or support (5 m across an open space) 4 - bedridden or wheelchair-bound 5 - ventilation assistance required (for any part of the day or night) 6 – dead |
|
Table 4:Medical Research Council (MRC) Scale for Manual Muscle Testing |
|
5 - patient can maintain position against maximal resistance and through the entire physiological range of motion of the joint 4 - patient can maintain position against moderate resistance, and moves actively through the entire physiological range of motion of the joint 3 - patient cannot maintain position against resistance, but can move the extremity against gravity through the full range of motion 2 - patient can move the extremity through part of the physiological range of motion if gravity is eliminated 1 - muscle contraction can be detected by palpation if gravity is eliminated 0 - no contractions identifiable
|
Table 5. The erasmus Guillain–Barré syndrome (GBS) Respiratory Insufficiency Score (eGRIS)
|
Measures |
Categories |
Score |
|
Days between onset of weakness and hospital admission |
<7 4-7 <3 |
0 1 2 |
|
Facial and/or bulbar weakness at hospital admission |
Absent Present |
0 1 |
|
MRC sum score at hospital admission |
60-51 50-41 40-31 30-21 <20 |
0 1 2 3 4 |
|
EGRIS |
NA |
0-7 |
NA, not applicable.
An eGRIS of 0–2 indicates a low risk of mechanical intervention, 3–4 indicates an intermediate risk of mechanical intervention and ≥5 indicates a high risk of mechanical intervention.
Table 6: The most important differential diagnoses of GBS
Intracranial
· Meningeosis neoplastica/leucaemica
· Brain stem encephalitis
Peripheral Nerves
· Axonal sub-acute recurrent neuropathy, with elevated CSF lactate levels in patients with a PDHcIa-mutation
· Metabolic disorders such as hypermagnesemia or hypophosphatemia
· Tick paralysis
· Heavy metal toxicity such as arsenic, gold and thallium
· Medication-induced neuropathy (e.g. vincristine, platinum compounds, nitrofurantoin, paclitaxel)
· Porphyria
· Critical illness neuropathy
· Vasculitis
· Diphtheria
Spinal cord
· Infarction, myelitis, compression
· Anterior horn motor neurons Polio and other enteroviruses that can trigger poliomyelitis, including West Nile Virus
Nerve roots
· Chronic inflammatory demyelinating polyneuropathy (CIDP)
· Cauda equina compression
Neuromuscular end-plate
· Myasthenia gravis
· Organophosphate poisoning
· Botulism
Muscle
· Critical illness
· Polymyositis
· Dermatomyositis
· Hypo-/hyperkalemia
· Periodic paralysis
- References
1- Sejvar JJ, Baughman AL, Wise M, Morgan OW. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology. 2011;36:123-133.
2- Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet.2016;388:717-727.
3- Asbury AK, Cornblath DR. Assessment of current diagnostic criteria for Guillain-Barre syndrome. Ann Neurol. 1990;27(S1):S21-S24.
4- F.G. Van der Meche, P.A. van Doorn, J. Meulstee, F.G. Jennekens, Diagnostic and classification criteria for the Guillain-Barre syndrome, Eur. Neurol. 45 (3)(2001) 133e139.
5- B.R. Wakerley, A. Uncini, N. Yuki, Guillain-Barre and Miller Fisher syndromes–new diagnostic classification, Nat. Rev. Neurol. 10 (9) (2014) 537e544.
6- Richard A.C. Hughes, A.V. Swan, van Doorn, A. Pieter, Intravenous immunoglobulin for Guillain-Barre syndrome, Cochrane Database Syst. Rev. (9) (2014), CD002063.
7- S. Chevret, R.A. Hughes, D. Annane, Plasma exchange for Guillain-Barre syndrome,
Cochrane Database Syst. Rev. 2 (2017), CD001798.
8- R. Korinthenberg, J.S. M€onting, Natural history and treatment effects in Guillain-Barr_e syndrome: a multicentre study, Arch. Dis. Child. 74 (4) (1996) 281e287.
9- Fokke C, van den Berg B, Drenthen J, Walgaard C, van Doorn PA, Jacobs BC. Diagnosis of Guillain-Barre syndrome and validation of Brighton criteria. Brain. 2014;137:33-43.
10- J. Roodbol, Y. de Wit, Marie-Claire, Bianca van den Berg, et al., Diagnosis of Guillain-Barre syndrome in children and validation of the Brighton criteria, J. Neurol. 264 (5) (2017) 856e861.
11- Kahlmann V, Roodbol J, van Leeuwen N, et al. Validated age-specific reference values for CSF total protein levels in children. Eur J Pae- diatr Neurol. 2017;21:654-660.
12- G.T. Doctor, S.K. Alexander, A. Radunovic, Guillain-Barre syndrome with exaggerated pleocytosis and anti-GM1 ganglioside antibodies, BMJ Case Rep. 2018 (2018).
13- Richard A.C. Hughes, D.R. Cornblath, Guillain-Barre syndrome, Lancet 366 (9497) (2005) 1653e1666.
14- Guo Y, Tian X, Wang X, Xiao Z. Adverse effects of immunoglobulin therapy. Front Immunol. 2018;9:1299.
15- Scottish Intercollegiate Guidelines Network (SIGN).
16- Diagnosis and treatment of Guillain-Barre Syndrome in childhood and adolescence: An evidence- and consensus-based guideline, 2020
17- European Academy of Neurology/Peripheral Nerve Society Guideline on diagnosis and treatment of Guillain–Barré syndrome, 2023
18- J.J. Sejvar, K.S. Kohl, J. Gidudu, et al., Guillain-Barre syndrome and Fisher syndrome: case definitions and guidelines for collection, analysis, and presentation of immunization safety data, Vaccine 29 (3) (2011) 599e612.
19- A. Uncini, S. Kuwabara, Electrodiagnostic criteria for Guillain-Barre syndrome:a critical revision and the need for an update, Clin. Neurophysiol. 123 (8)(2012) 1487e1495.
20- Van den Bergh, Y.K. Peter, F. Pi_eret, J.L. Woodard, et al., Guillain-Barr_e Syndrome Subtype Diagnosis: A Prospective Multicentric European Study, Muscle Nerve, 2018.
21- Umapathi T, Lim CSJ, Ng BCJ, Goh EJH, Ohnmar O. A simplified, graded, electrodiagnostic criterion for Guillain-Barre syndrome that incorporates sensory nerve conduction studies. Sci Rep. 2019;9:7724
22- Derksen A, Ritter C, Athar P, et al. Sural sparing pattern discriminates Guillain-Barre syndrome from its mimics. Muscle Nerve. 2014; 50:780-784.
23- Coskun A, Kumandas S, Pac A, Karahan OI, Gulec M, Baykara M. Childhood Guillain-Barre syndrome. MR imaging in diagnosis and follow-up. Acta Radiol. 2003;44:230-235.
24- Gorson KC, Ropper AH, Muriello MA, Blair R. Prospective evaluation of MRI lumbosacral nerve root enhancement in acute Guillain-Barre syndrome. Neurology. 1996;47:813-817.
25- Mulkey SB, Glasier CM, El-Nabbout B, et al. Nerve root enhancement on spinal MRI in pediatric Guillain-Barre syndrome. Pediatr Neurol. 2010;43:263-269.
26- Yikilmaz A, Doganay S, Gumus H, Per H, Kumandas S, Coskun A. Magnetic resonance imaging of childhood Guillain-Barre syndrome. Childs Nerv Syst. 2010;26:1103-1108.
27- Jacobs, B. C. et al. The spectrum of antecedent infections in Guillain- Barré syndrome: a case- control study. Neurology 51, 1110–1115 (1998)
28- Cao- Lormeau, V. M. et al. Guillain- Barré syndrome outbreak associated with Zika virus infection in French Polynesia: a case- control study. Lancet 387, 1531–1539 (2016).
29- Van den Berg, B. et al. Guillain- Barré syndrome associated with preceding hepatitis E virus infection. Neurology 82, 491–497 (2014).
30- Patone M, Handunnetthi L, Saatci D, et al. Neurological complications after first dose of COVID- 19 vaccines and SARS- CoV- 2 infection. Nat Med. 2021;27:2144-2153.
31- Keddie S, Pakpoor J, Mousele C, et al. Epidemiological and cohort study finds no association between COVID- 19 and Guillain- Barre syndrome. Brain. 2021;144:682-693.
32- Martin Arias LH, Sanz R, Sainz M, Treceno C, Carvajal A. Guillain- Barre syndrome and influenza vaccines: a meta- analysis. Vaccine. 2015;33:3773-3778.
33- N. Andrews, J. Stowe, R. Al-Shahi Salman, E. Miller, Guillain-Barre syndrome and H1N1 (2009) pandemic influenza vaccination using an AS03 adjuvanted vaccine in the United Kingdom: self-controlled case series, Vaccine 29 (45) (2011) 7878e7882.
34- P. Haber, J. Sejvar, Y. Mikaeloff, F. DeStefano, Vaccines and guillain-barre syndrome, Drug Saf. 32 (4) (2009) 309e323.
35- Hashemilar M, Barzegar M, Nikanfar M, et al. Evaluating the status of antiganglioside antibodies in children with Guillain-Barre syndrome. Neuroimmunomodulation. 2014;21:64-68.
36- Gregson NA, Koblar S, Hughes RA. Antibodies to gangliosides in Guillain-Barre syndrome: specificity and relationship to clinical features. Q J Med. 1993;86:111-117.
37- Caudie C, Quittard Pinon A, Taravel D, et al. Preceding infections and anti-ganglioside antibody profiles assessed by a dot immunoassay in 306 French Guillain-Barre syndrome patients. J Neurol. 2011; 258:1958-1964.
38- Carpo M, Pedotti R, Lolli F, et al. Clinical correlate and fine specificity of anti-GQ1b antibodies in peripheral neuropathy. J Neurol Sci.1998;155:186-191.
39- Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serumanti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barre syndrome: clinical and immunohistochemical studies. Neurology. 1993;43:1911-1917.
40- R. Korinthenberg, J. Schessel, J. Kirschner, J.S. Monting, Intravenously administered immunoglobulin in the treatment of childhood Guillain-Barre syndrome: a randomized trial, Pediatrics 116 (1) (2005) 8-14.
41- D. Singh-Grewal, A. Kemp, M. Wong, A prospective study of the immediate and delayed adverse events following intravenous immunoglobulin infusions, Arch Dis Child. 91 (8) (2006) 651 – 654.
42- Verboon, C. ∙ van Doorn ∙ Pieter, A.Treatment dilemmas in Guillain-Barre syndrome.J. Neurol. Neurosurg. Psychiatry. 2017; 88:346-352
43- S. Chevret, R.A. Hughes, D. Annane, Plasma exchange for Guillain-Barre syndrome, Cochrane Database Syst. Rev. 2 (2017), CD001798.
44- Hughes RA, Newsom-Davis JM, Perkin GD, Pierce JM. Controlled trial prednisolone in acute polyneuropathy. Lancet. 1978;2:750-753.
45- RA Hughes, RAC, Cornblath DR. Guillain-Barre syndrome. Lancet 2005;366 (9497): 1653-66.
46- Hughes RA, Brassington R, Gunn AA, van Doorn PA. Corticosteroids for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2016;10: CD001446.
47- Guillain-Barre Syndrome Steroid Trial Group. Double-blind trial of intravenous methylprednisolone in Guillain-Barre syndrome. Lancet 1993;341:586-590.
48- van Koningsveld R, Schmitz PI, Meche FG, Visser LH, Meulstee J,Van Doorn PA. Effect of methylprednisolone when added to standard treatment with intravenous immunoglobulin for Guillain-Barre syndrome: randomised trial. Lancet. 2004;363:192-196.
49- Kleyweg RP, van der Meche FG. Treatment related fluctuations in Guillain-Barre syndrome after high-dose immunoglobulins or plasma-exchange. J Neurol Neurosurg Psychiatry. 1991;54:957-960.
50- Hughes RA, Swan AV, Raphael JC, Annane D, van Koningsveld R, van Doorn PA. Immunotherapy for Guillain-Barre syndrome: a systematic review. Brain. 2007;130:2245-2257.
51- Van den Bergh PYK, van Doorn PA, Hadden RDM, et al. European Academy of Neurology/Peripheral Nerve Society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint Task Force-second revision.J Peripher Nerv Syst. 2021;26:242-268.
52- Ruts L, Drenthen J, Jacobs BC, van Doorn PA. Distinguishing acute onset CIDP from fluctuating Guillain-Barre syndrome: a prospective study. Neurology. 2010;74:1680-1686
53- Halstead SK, Zitman FM, Humphreys PD, et al. Eculizumab prevents anti-ganglioside antibody-mediated neuropathy in a murine model. Brain. 2008;131:1197-1208.
54- Davidson AI, Halstead SK, Goodfellow JA, et al. Inhibition of complement in Guillain-Barre syndrome: the ICA-GBS study. J Peripher Nerv Syst. 2017;22:4-12.
55- Misawa S, Kuwabara S, Sato Y, et al. Safety and efficacy of eculizumab in Guillain-Barre syndrome: a multicentre, double-blind, randomized phase 2 trial. Lancet Neurol. 2018;17:519-529.
56- van Koningsveld R, Steyerberg EW, Hughes RA, Swan AV, van Doorn PA, Jacobs BC. A clinical prognostic scoring system for Guillain-Barre syndrome. Lancet Neurol. 2007;6:589-594.
57- Walgaard C, Lingsma HF, Ruts L, van Doorn PA, Steyerberg EW, Jacobs BC. Early recognition of poor prognosis in Guillain-Barre syndrome. Neurology. 2011;76:968-975.
58- Alessandro L, Pastor Rueda JM, Wilken M, et al. Differences between acute-onset chronic inflammatory demyelinating polyneuropathy and acute inflammatory demyelinating polyneuropathy in adult patients. J Peripher Nerv Syst. 2018;23:154-158.
59- Dionne A, Nicolle MW, Hahn AF. Clinical and electrophysiological parameters distinguishing acute-onset chronic inflammatory demyelinating polyneuropathy from acute inflammatory demyelinating polyneuropathy. Muscle Nerve. 2010;41:202-207.
60- Grimm A, Oertl H, Auffenberg E, et al. Differentiation between Guillain-Barre syndrome and acute-onset chronic inflammatory demyelinating polyradiculoneuritis: a prospective follow-up study using ultrasound and neurophysiological measurements. Neurotherapeutics. 2019;16:838-847.
61- Vural A, Doppler K, Meinl E. Autoantibodies against the node of Ranvier in seropositive chronic inflammatory demyelinating polyneuropathy: diagnostic, pathogenic, and therapeutic relevance. Front Immunol. 2018;9:1029.
62- Y. Nevo, H. Topaloglu, 88th ENMC international workshop: childhood chronic inflammatory demyelinating polyneuropathy (including revised diagnostic criteria), Naarden, The Netherlands, December 8-10, Neuromuscul. Disord. 12(2) (2000) 195e200, 2002.