
كتاب
Diagnosis and management of Guillian Barre Syndrome in children and adolescents
متطلبات الإكمال
"last update: 3 Nov 2025" Download Guideline
- Introduction
Guillain Barre syndrome (GBS) is the most common cause of acute flaccid paralysis with an annual global incidence of approximately
1–2 per 100 000 person years(1,2) The diagnosis of GBS relies upon a combination of clinical features, often with support of electrodiagnostic and laboratory features. Most diagnostic criteria for GBS require a combination of history, neurological examination, cerebrospinal fluid (CSF)and electrodiagnostic results.(3)
GBS can be subclassified into a number of variants, depending on clinical presentation and electrophysiological findings (4) .The classic variants are acute demyelinating inflammatory polyneuropathy (AIDP) and acute motor axonal neuropathy (AMAN).
These are characterized by rapidly-progressing, ascending symmetrical weakness, with attenuation or loss of muscle proprioceptive reflexes. Among pediatric patients in the peak phase of the disease, 75% can no longer walk unaided, 30% are tetraparetic,35-50% show cranial nerve involvement, and 15-20% have respiratory failure and/or autonomic dysfunction. Furthermore, up to 70% suffer from neuropathic pain, which can be severe and occasionally occur as the first symptom. Localized variants of GBS include Miller Fisher Syndrome (MFS) (cranial nerve affection and ataxia, areflexia, serum anti-GQ1b antibody detection in >90% cases) and the pharyngeal-cervical-brachial variant (predominantly bulbar and neck weakness, serum IgG antibodies against GT1a frequently detected), both of which are extremely rare in childhood (5) .Immunomodulatory therapy with plasmapheresis (PE) or intravenous immunoglobulins (IVIG) has been proven effective in GBS namely by accelerating the improvement of weakness. (6,7)
The improvement follows a plateau phase that ranges from days to weeks and runs an extremely variable course, regardless of whether a specific therapy has been applied or not. The overall long-term prognosis for children with GBS is more favorable than that in adults, whereby the majority of children largely regain motor function.(8)
There are several forms of GBS within the disease spectrum, which are defined by the clinical features, with some overlap: fig 1
1- Motor-sensory((weakness with sensory symptoms and/or signs) and motor ((weakness without sensory symptoms or signs )GBS are considered ‘typical’ GBS
2- Pharyngeal–cervical–brachial GBS with weakness in the corresponding regions, bilateral facial weakness with limb paraesthesias (but no limb weakness)
3- Paraparetic variant with weakness starting in the legs that may evolve to tetraparesis
4- Pure sensory syndrome of numbness or tingling, reduced or absent reflexes, and sometimes ataxia or pseudoathetosis, but no weakness, with a time course like GBS. Nerve conduction in these patients may show signs of demyelination in sensory or motor nerves. Although the patients with this pure sensory syndrome do not fulfil all diagnostic criteria for GBS, this probably represents a rare sensory variant of GBS
5- MFS spectrum : Patients with typical MFS have ophthalmoplegia, ataxia and areflexia
Bickerstaff brainstem encephalitis (BBE) is considered a rare variant of MFS that clinically has a combination of ophthalmoplegia, ataxia, pyramidal tract signs and impaired consciousness.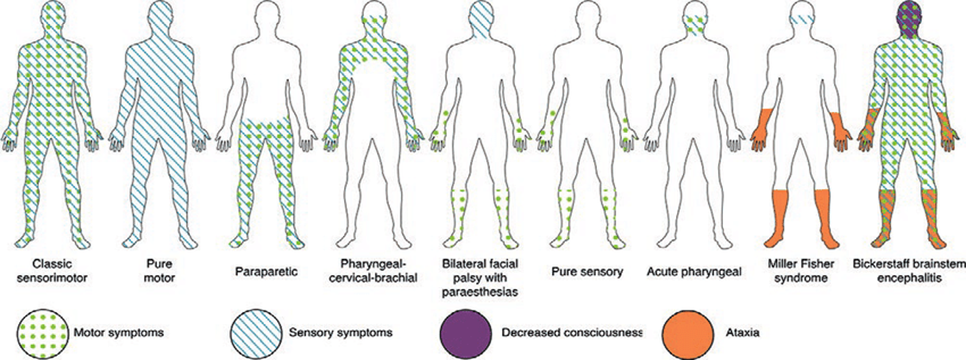
Fig 1: Forms of GBS
